UShER
UShER is a program for rapid, accurate placement of samples to existing phylogenies. It is available for downloading here and is updated regularly. While not restricted to SARS-CoV-2 phylogenetic analyses, it has enabled real-time phylogenetic analyses and genomic contact tracing in that its placement is orders of magnitude faster and more memory-efficient than previous methods, and is being widely used by several SARS-CoV-2 research groups, including the UCSC Genome Browser team and Rob Lanfear’s global phylogeny releases. We recommend using Taxonium (formerly known as Cov2Tree or Taxodium) developed by Theo Sanderson to visualize these trees.
Methodology
Given existing samples, whose genotypes and phylogenetic tree is known, and the genotypes of new samples, UShER aims to incorporate new samples into the phylogenetic tree while preserving the topology of existing samples and maximizing parsimony. UShER’s algorithm consists of two phases: (i) the pre-processing phase and (ii) the placement phase.
Pre-processing
In the pre-processing phase, UShER accepts the phylogenetic tree of existing samples in a Newick format and their genotypes, specified as a set of single-nucleotide variants with respect to a reference sequence (UShER currently ignores indels), in a VCF format. For each site in the VCF, UShER uses the Fitch-Sankoff algorithm to find the most parsimonious nucleotide assignment for every node of the tree (UShER automatically labels internal tree nodes). When a sample contains ambiguous genotypes, multiple nucleotides may be most parsimonious at a node. To resolve these, UShER assigns it any one of the most parsimonious nucleotides with preference, when possible, given to the reference base. UShER also allows the VCF to specify ambiguous bases in samples using IUPAC format, which are also resolved to a unique base using the above strategy. When a node is found to carry a mutation, i.e. the base assigned to the node differs from its parent, the mutation gets added to a list of mutations corresponding to that node. Finally, UShER uses protocol buffers to store in a file, the Newick string corresponding to the input tree and a list of lists of node mutation, which we refer to as mutation-annotated tree object, as shown in the figure below.
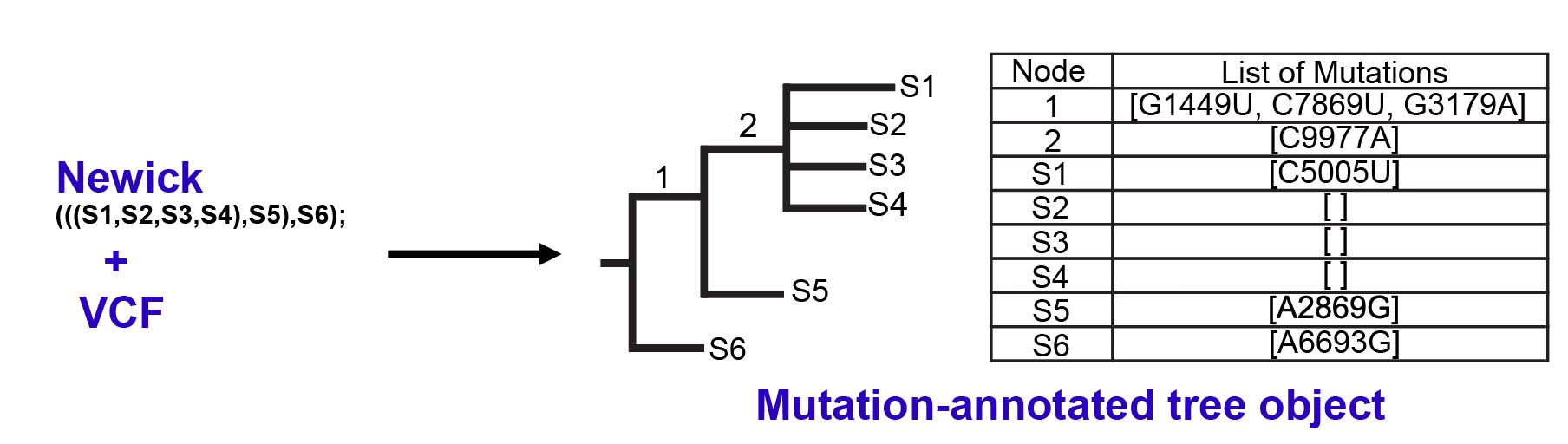
The mutation-annotated tree object carries sufficient information to derive parsimony-resolved genotypes for any tip of the tree using the sequence of mutations from the root to that tip. For example, in the above figure, S5 can be inferred to contain variants G1149U, C7869U, G3179A and A2869G with respect to the reference sequence. Compared to other tools that use full multiple-sequence alignment (MSA) to guide the placement, UShER’s mutation-annotated tree object is compact and is what helps make it fast.
Placement
In the placement phase, UShER loads the pre-processed mutation-annotated tree object and the genotypes of new samples in a VCF format and sequentially adds the new samples to the tree. For each new sample, UShER computes the additional parsimony score required for placing it at every node in the current tree while considering the full path of mutations from the root of the tree to that node. Next, UShER places the new sample at the node that results in the smallest additional parsimony score. When multiple node placements are equally parsimonious, UShER picks the node with a greater number of descendant leaves for placement. If the choice is between a parent and its child node, the parent node would always be selected by this rule. However, a more accurate placement should reflect the number of leaves uniquely attributable to the child versus parent node. Therefore, in these cases, UShER picks the parent node if the number of descendant leaves of the parent that are not shared with the child node exceed the number of descendant leaves of the child. The figure below shows a new sample, S7, containing variants G1149U and C9977A being added to the previous mutation-annotated tree object in a parsimony-optimal fashion (with a parsimony score of 1 for the mutation C9977A). UShER also automatically imputes and reports ambiguous genotypes for the newly added samples and ignores missing bases, such as ‘N’ or ‘.’ (i.e. missing bases never contribute to the parsimony score).
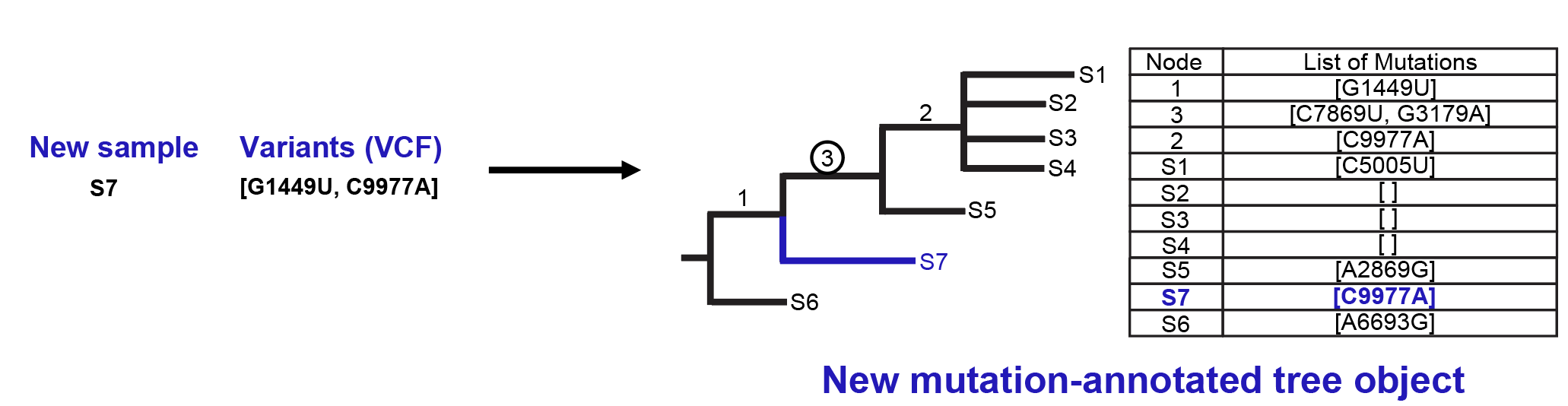
At the end of the placement phase, UShER allows the user to create another protocol-buffer (protobuf) file containing the mutation-annotated tree object for the newly generated tree including added samples as also shown in the example figure above. This allows for another round of placements to be carried out over and above the newly added samples.
Options
--vcf (-v): Input VCF file (in uncompressed or gzip-compressed .gz format). (REQUIRED)
--tree (-t): Input tree file.
--outdir (-d): Output directory to dump output and log files.
--load-mutation-annotated-tree (-i): Load mutation-annotated tree object.
--save-mutation-annotated-tree (-o): Save output mutation-annotated tree to the specified filename.
--sort-before-placement-1 (-s): Sort new samples based on computed parsimony score and then number of optimal placements before the actual placement (EXPERIMENTAL).
--sort-before-placement-2 (-S): Sort new samples based on the number of optimal placements and then the parsimony score before the actual placement (EXPERIMENTAL).
--reverse-sort (-r): Reverse the sorting order of sorting options (sort-before-placement-1 or sort-before-placement-2). (EXPERIMENTAL)
--collapse-tree (-c): Collapse internal nodes of the input tree with no mutations and condense identical sequences in polytomies into a single node and the save the tree to file condensed-tree.nh in outdir.
--max-uncertainty-per-sample (-e): Maximum number of equally parsimonious placements allowed per sample beyond which the sample is ignored. Default = 1000000.
--write-uncondensed-final-tree (-u): Write the final tree in uncondensed format and save to file uncondensed-final-tree.nh in outdir.
--write-subtrees-size (-k): Write minimum set of subtrees covering the newly added samples of size equal to or larger than this value. Default = 0.
--write-parsimony-scores-per-node (-p): Write the parsimony scores for adding new samples at each existing node in the tree without modifying the tree in a file names parsimony-scores.tsv in outdir.
--multiple-placements (-M): Create a new tree up to this limit for each possibility of parsimony-optimal placement. Default = 1.
--retain-input-branch-lengths (-l): Retain the branch lengths from the input tree in out newick files instead of using number of mutations for the branch lengths.
--threads (-T): Number of threads to use when possible. Default = use all available cores.
--help (-h): Print help messages.
Usage
Display help message
To familiarize with the different command-line options of UShER, it would be useful to view its help message using the command below:
usher --help
Pre-processing global phylogeny
The following example command pre-processes the existing phylogeny (global_phylo.nh) and using the genotypes (global_samples.vcf) and generates the mutation-annotated tree object that gets stored in a protobuf file (global_assignments.pb). Note that UShER would automatically place onto the input global phylogeny any samples in the VCF (to convert a fasta sequence to VCF, consider using Fasta2USHER that are missing in the input global phylogeny using its parsimony-optimal placement algorithm. This final tree is written to a file named final-tree.nh in the folder specified by --outdir or -d option (if not specified, default uses current directory).
usher -t test/global_phylo.nh -v test/global_samples.vcf -o global_assignments.pb -d output/
By default, UShER uses all available threads but the user can also specify the number of threads using the --threads or -T command-line parameter.
UShER also allows an option during the pre-processing phase to collapse nodes (i.e. delete the node after moving its child nodes to its parent node) that are not inferred to contain a mutation through the Fitch-Sankoff algorithm as well as to condense nodes that contain identical sequences into a single representative node. This is the recommended usage for UShER as it not only helps in significantly reducing the search space for the placement phase but also helps reduce ambiguities in the placement step and can be done by setting the --collapse-tree or -c parameter. The collapsed input tree is stored as condensed-tree.nh in the output directory.
usher -t test/global_phylo.nh -v test/global_samples.vcf -o global_assignments.pb -c -d output/
Note the the above command would condense identical sequences, namely S2, S3 and S4, in the example figure above into a single condensed new node (named something like node_1_condensed_3_leaves). If you wish to display the collapsed tree without condensing the nodes, also set the --write-uncondensed-final-tree or -u option, for example, as follows:
usher -t test/global_phylo.nh -v test/global_samples.vcf -o global_assignments.pb -c -u -d output/
The above commands saves the collapsed but uncondensed tree as uncondensed-final-tree.nh in the output directory.
Placing new samples
Once the pre-processing is complete and a mutation-annotated tree object is generate (e.g. global_assignments.pb), UShER can place new sequences whose variants are called in a VCF file (e.g. new_samples.vcf) to existing tree as follows:
usher -i global_assignments.pb -v test/new_samples.vcf -u -d output/
Again, by default, UShER uses all available threads but the user can also specify the number of threads using the –threads command-line parameter.
The above command not only places each new sample sequentially, but also reports the parsimony score and the number of parsimony-optimal placements found for each added sample. UShER displays warning messages if several (>=4) possibilities of parsimony-optimal placements are found for a sample. This can happen due to several factors, including (i) missing data in new samples, (ii) presence of ambiguous genotypes in new samples and (iii) structure and mutations in the global phylogeny itself, including presence of multiple back-mutations.
In addition to the global phylogeny, one often needs to contextualize the newly added sequences using subtrees of closest N neighbouring sequences, where N is small. UShER allows this functionality using --write-subtrees-size or -k option, which can be set to an arbitrary N, such as 20 in the example below:
usher -i global_assignments.pb -v test/new_samples.vcf -u -k 20 -d output/
The above command writes subtrees to files names subtree-<subtree-number>.nh. It also write a text file for each subtree (named subtree-<subtree-number>-mutations.txt showing mutations at each internal node of the subtree. If the subtrees contain condensed nodes, it writes the expanded leaves for those nodes to text files named subtree-<subtree-number>-expanded.txt.
Finally, the new mutation-annotated tree object can be stored again using --save-mutation-annotated-tree or -o option (overwriting the loaded protobuf file is allowed).
usher -i global_assignments.pb -v test/new_samples.vcf -u -o new_global_assignments.pb -d output/
Features
In addition to simply placing samples on an existing phylogeny, UShER provides the user with several points of additional information, including measurements of uncertainty in sample placement, and is capable of auxiliary analyses:
Branch Parsimony Score
UShER also allows quantifying the uncertainty in placing new samples by reporting the parsimony scores of adding new samples to all possible nodes in the tree without actually modifying the tree (this is because the tree structure, as well as number of possible optimal placements could change with each new sequential placement). In particular, this can help the user explore which nodes of the tree result in a small and optimal or near-optimal parsimony score. This can be done by setting the --write-parsimony-scores-per-node or -p option, for example, as follows:
usher -i global_assignments.pb -v test/new_samples.vcf -p -d output/
The above command writes a file parsimony-scores.tsv containing branch parsimony scores to the output directory. Note that because the above command does not perform the sequential placement on the tree, the number of parsimony-optimal placements reported for the second and later samples could differ from those reported with actual placements.
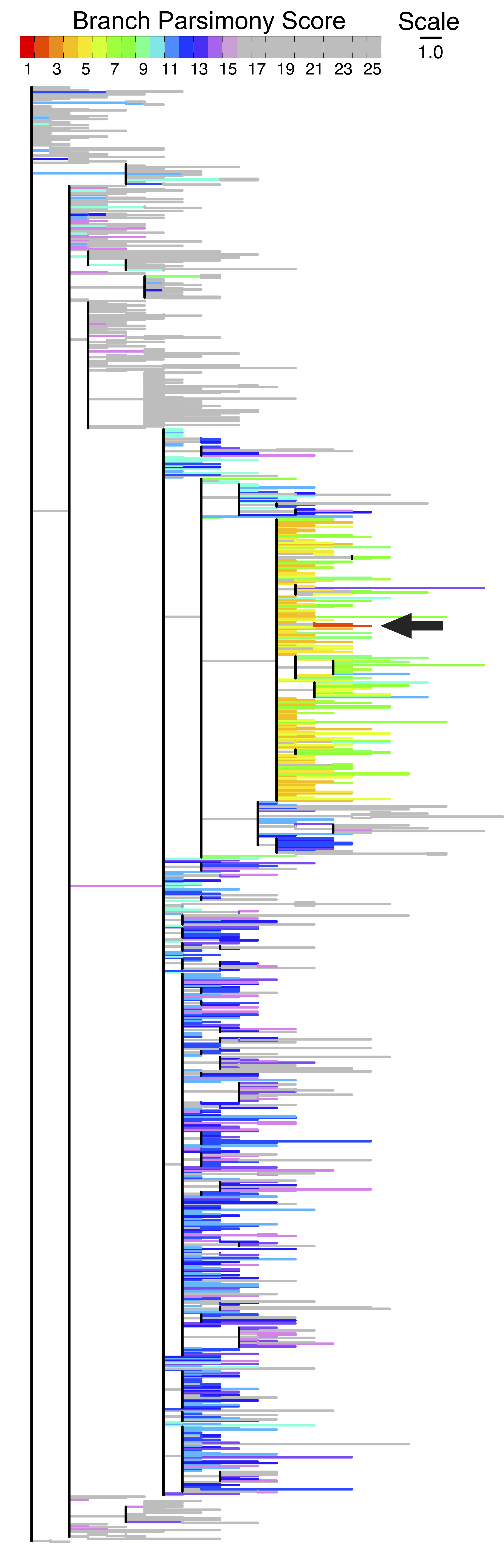
The figure below shows how branch parsimony score could be useful for uncertainty analysis. The figure shows color-coded parsimony score of placing a new sample at different branches of the tree with black arrow pointing to the branch where the placement is optimal. As can be seen from the color codes, the parsimony scores are low (implying good alternative placement) for several neighboring branches of the optimal branch.
Multiple parsimony-optimal placements
To further aid the user to quantify phylogenetic uncertainty in placement, UShER has an ability to enumerate all possible topologies resulting from equally parsimonious sample placements. UShER does this by maintaining a list of mutation-annotated trees (starting with a single mutation-annotated tree corresponding to the input tree of existing samples) and sequentially adds new samples to each tree in the list while increasing the size of the list as needed to accommodate multiple equally parsimonious placements for a new sample. This feature is available using the --multiple-placements or -M option in which the user specifies the maximum number of topologies that UShER should maintain before it reverts back to using the default tie-breaking strategy for multiple parsimony-optimal placements in order to keep the runtime and memory usage of UShER reasonable.
usher -i global_assignments.pb -v <USER_PROVIDED_VCF> -M -d output/
Note that if the number of equally parsimonious placements for the initial samples is large, the tree space can get too large too quickly and slow down the placement for the subsequent samples. Therefore, UShER also provides an option to sort the samples first based on the number of equally parsimonious placements using the -S option.
usher -i global_assignments.pb -v <USER_PROVIDED_VCF> -M -S -d output/
There are many ways to interpret and visualize the forest of trees produced by multiple placements. One method is to use DensiTree, as shown using an example figure (generated using the phangorn package) below:
Updating multiple input trees
UShER is also fast enough to allow users to update multiple input trees incorporating uncertainty in tree resonstruction, such as multiple bootstrap trees. While we do not provide an explicit option to input multiple trees at once, UShER can be run independently for each input tree and place new samples. We recommend the user to use the GNU parallel utility to do so in parallel using multiple CPU cores while setting -T 1 for each UShER task.
Finding sister clades
To determine the accuracy of each sample placement, one might be interested in knowing all of the sister clades of that sample on the final tree. We provide a utility for this calculation here. find_sister_clades takes the following options:
--tree: Input tree file.
--samples: File containing missing samples.
--generations: Number of generations.
--help: Print help messages.
An example usage of this function is given below:
find_sister_clades --generations 1 final-tree.nh --samples list-of-samples.txt > sister_clades.txt
The samples file should have the name of each sample of interest exactly as it appears in the input tree file. The output file will contain the name of each sample with a :, followed by each sister sample at the input generation, separated by a new-line character. Lists of sister clades for each sample are separated by two new-lines characters.
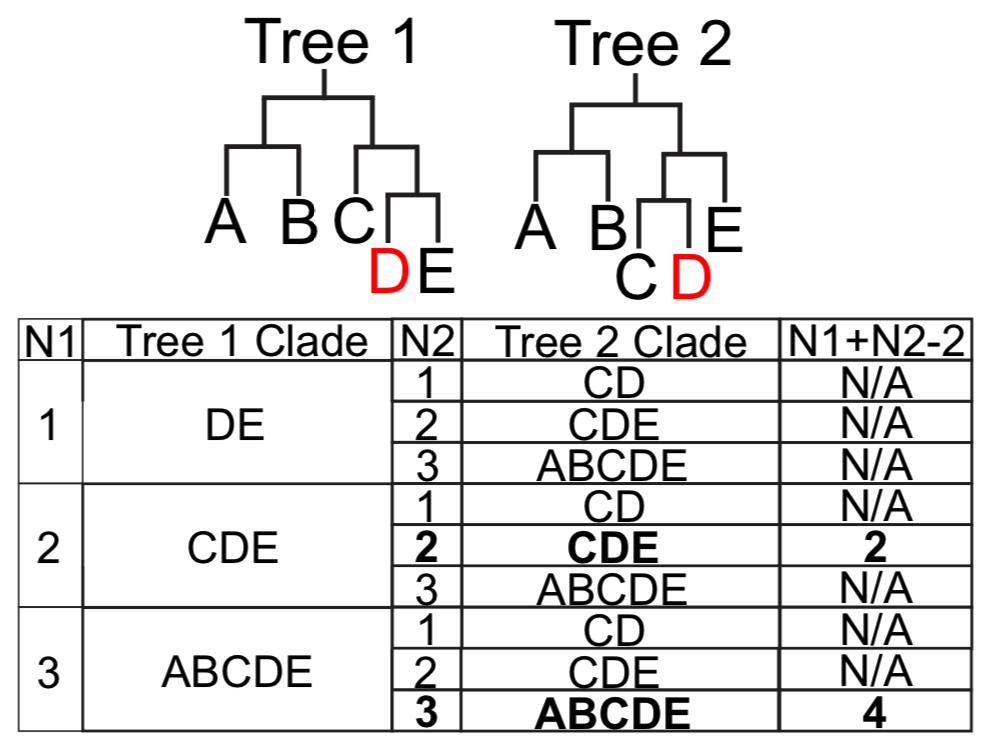
We also described a method for measuring tree congruence involving comparing the sister clades at several generations, and finding the minimum combined number of generations at which a given sample has the same sister clades in two trees. For clarity, we provide the figure below:
Converting raw sequences into VCF for UShER input
We provide instructions below for converting raw genomic sequences in fasta format into VCF for UShER’s input.
Generating Multiple Sequence Alignment (MSA)
Users can generate multiple sequence alignment of the input sequences using MAFFT that is already installed with UShER package. For example, we provide a number of example SARs-CoV-2 sequences in test/Fasta2UShER that can be combined in a single fasta file called combined.fa and aligned together using the SARS-CoV-2 reference sequence test/NC_045512v2.fa as follows:
cat ./test/Fasta2UShER/* > ./test/combined.fa
mafft --thread 10 --auto --keeplength --addfragments ./test/combined.fa ./test/NC_045512v2.fa > ./test/myAlignedSequences.fa
If you have a large number of sequences, we recommend using Robert Lanfear’s global_profile_alignment.sh, which can reduce memory requirements by splitting alignments and performing them in parallel.
Converting MSA to VCF
Users can then use the tool faToVcf, which is also installed via UShER’s package, to then convert the fasta file containing multiple alignment of input sequences into a VCF. If the file ./test/myAlignedSequences.fa includes the reference sequence (for SARS-CoV-2, NC_045512.2) as its first sequence, then faToVcf can be run like this:
faToVcf ./test/myAlignedSequences.fa ./test/test_merged.vcf
If the reference sequence is included in ./test/myAlignedSequences.fa but is not the first sequence, then its name needs to be specified using the -ref=... option:
faToVcf -ref=NC_045512.2 ./test/myAlignedSequences.fa ./test/test_merged.vcf
If the reference sequence is not included in ./test/myAlignedSequences.fa then it can be added in the bash shell using a named pipe like this:
faToVcf <(cat NC_045512.2.fa ./test/myAlignedSequences.fa) ./test/test_merged.vcf
For SARS-CoV-2 data, we recommend downloading problematic_sites_sarsCov2.vcf and using it for masking problematic sites as follows:
wget https://raw.githubusercontent.com/W-L/ProblematicSites_SARS-CoV2/master/problematic_sites_sarsCov2.vcf
faToVcf -maskSites=problematic_sites_sarsCov2.vcf ./test/myAlignedSequences.fa ./test/test_merged_masked.vcf
The resulting VCF files test_merged.vcf and test_merged_masked.vcf from the above commands should be compatible with UShER.
Presentations
Russ Corbett-Detig has created a module on UShER for the CDC:
Yatish Turakhia has presented on UShER at the Covid-19 Dynamics & Evolution Meeting, held virtually on October 19-20, 2020. You can find his slides here.
Publications
Turakhia Y, Thornlow B, Hinrichs A, De Maio N, Gozashti L, Lanfear R, Haussler D, and Corbett-Detig R. Ultrafast Sample placement on Existing tRees (UShER) enables real-time phylogenetics for the SARS-CoV-2 pandemic., Nature Genetics. 2021. 1-8.